Institutional Review Boards: Actions Needed to Improve Federal Oversight and Examine Effectiveness
Fast Facts
Institutional Review Boards (IRBs) assess the ethics and safety of research studies involving human subjects, such as behavioral studies or clinical trials for new drugs or medical devices.
Health and Human Services oversees about 2,300 U.S.-based IRBs through routine or for-cause inspections to assess if they are following federal laws when reviewing research. But few IRBs are inspected. For example, one HHS agency aims to do just 3-4 routine inspections each year. Also, HHS agencies haven't examined how many inspections are needed or if inspections could be changed to further reduce risks to human subjects.
Our recommendations address this.

Highlights
What GAO Found
Institutional review boards (IRB) are groups that review ethical and safety considerations for research involving human subjects, such as clinical trials.
General Institutional Review Board (IRB) Process
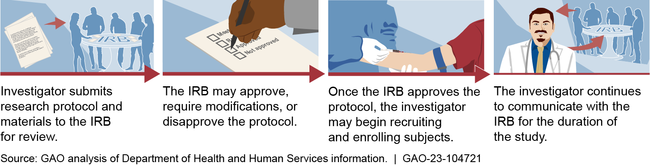
Most IRBs are based at universities, according to Department of Health and Human Services (HHS) data. University-based IRBs were also responsible for reviewing most research involving certain investigational drugs from calendar years 2012 through 2020, according to Food and Drug Administration (FDA) data. Some IRBs are independent, meaning they are not part of institutions that conduct or sponsor research. FDA data show these independent IRBs have reviewed an increasing share of investigational drug research: 25 percent of this research in 2012, and 48 percent in 2021. At the same time, the number of independent IRBs has decreased largely due to consolidation; this is, in part, related to private equity investment in IRBs.
FDA and HHS's Office for Human Research Protections (OHRP) oversee about 2,300 U.S.-based IRBs (operated by about 1,800 separate organizations, which may register and operate one or more IRB) through routine or for-cause inspections. These inspections assess whether IRBs follow federal regulations when reviewing research. FDA and OHRP consider several factors when selecting organizations for inspections, such as the volume of research reviewed. However, GAO found the agencies inspect relatively few IRBs. OHRP officials said they aim to conduct three to four routine inspections annually, while FDA conducted an average of 133 inspections annually between fiscal years 2010 and 2021. Neither agency has conducted a risk-based assessment of their IRB inspection program to help ensure they inspect enough IRBs annually and to optimize their responsibilities in protecting human subjects. Such an approach would be consistent with federal risk management principles.
While the agencies oversee IRBs to determine their adherence to regulations, OHRP and FDA have not assessed to what extent IRB reviews are effective in protecting human subjects. This is because the agencies have not determined the best approaches for doing so. Evaluating effectiveness is challenging in part due to an absence of validated measures and because IRBs are only one part of the framework of stakeholders responsible for protecting human subjects. Convening stakeholders to identify approaches for evaluating IRB effectiveness would be consistent with OHRP and FDA responsibilities and change management practices, and would help provide assurance that IRBs are successful in protecting human subjects.
Why GAO Did This Study
IRBs review research studies involving human subjects to ensure that risks to subjects are minimized and participants have sufficient information to consent to participate. In the past, IRBs were based at research institutions, such as academic centers. Over time, independent IRBs have played a more prominent role in reviewing research on human subjects. Some policymakers and others have raised questions about the increased use of independent IRBs and the effects on protecting human subjects.
GAO was asked to examine independent IRBs, processes used to protect human subjects, and standards of IRB quality, among other things. This report describes the composition of the IRB market and examines OHRP and FDA oversight of IRBs, among other objectives.
GAO reviewed federal laws and regulations and articles published between 2010 and June 2021; analyzed IRB registration, drug application, and inspection data; and interviewed FDA and OHRP officials, experts and stakeholders, and 11 IRBs selected for variation in type, size, and other factors.
Recommendations
GAO is making four recommendations, including that HHS and FDA conduct annual risk assessments to determine if the agencies are routinely inspecting an adequate number of IRBs and to optimize the use of inspections in the oversight of IRBs and protection of research participants, and examine and implement approaches for measuring IRB effectiveness. HHS concurred with the recommendations.
Recommendations for Executive Action
| Agency Affected | Recommendation | Status |
|---|---|---|
| Department of Health and Human Services | The Assistant Secretary for Health should ensure that OHRP takes steps to ensure the accuracy of protocol data collected in OHRP's IRB registry. This could include updating instructions to IRBs and examining data accuracy for a sample of IRBs. (Recommendation 1.) |
In August 2023, HHS reported steps OHRP was taking to address this recommendation. HHS noted OHRP was working to modify its IRB registration system to prompt IRBs applying for, renewing, or updating their OHRP registration to provide accurate information about the number of protocols they review. Additionally, HHS reported OHRP planned to evaluate its IRB registration instructions as part of the next Paperwork Reduction Act Information Collection Request renewal. In 2024, OHRP described the importance of reporting accurate IRB registration information in a publication sponsored by the National Council of University Research Administrators. In it, OHRP also communicated changes the agency planned to make to IRB registration instructions that may help institutions provide more accurate protocol data. HHS also noted that OHRP was considering ways to obtain information that could help to address this recommendation during its IRB inspections. In December 2025, HHS stated that OHRP was continuing work to modernize its IRB registration system, update FAQs and training tools, and explore options to use IRB data it collects as part of a risk-based analysis to select inspection sites. For example, HHS reported OHRP had updated the IRB application form and instructions and was awaiting leadership review, which is the first of several steps needed to improve the accuracy and completeness of protocol information collected through the IRB registration system. OHRP said staffing reductions impeded their progress towards this recommendation. We will update the status of this recommendation when we receive additional information on the actions OHRP has completed to help ensure the accuracy of the protocol data it collects.
|
| Department of Health and Human Services | The Assistant Secretary for Health should ensure that OHRP conducts an annual risk assessment to determine whether the agency is conducting an adequate number of routine IRB inspections and to optimize the use of IRB inspections in the oversight of IRBs and protection of research participants. (Recommendation 2) |
In August 2023, HHS reported that OHRP was taking steps to address this recommendation, including by considering options for conducting an annual risk assessment and discussing approaches with FDA and other federal entities that support or conduct human subjects. In April 2024, HHS reiterated the steps it was taking to address this recommendation, noting that identifying which IRBs to inspect will be a complex process that will likely take 2-3 years to successfully implement. In December 2025, HHS reported OHRP was developing a new approach for selecting IRBs for inspection and had examined sources of data to understand IRBs that oversee the highest volume of HHS-supported or conducted research and may be more vulnerable to risk of noncompliance with HHS regulations. HHS noted OHRP is also working to create a dashboard that integrates data from OHRP's internal systems with NIH funding data to evaluate patterns of complaints and reportable events. OHRP plans to integrate this dashboard in its process for making decisions on not-for-cause evaluations. HHS also noted that reductions in OHRP staff and competing priorities will affect their progress in addressing the recommendation. We will update the status of this recommendation when we receive additional information regarding actions OHRP has completed to conduct this assessment.
|
| Food and Drug Administration | The Commissioner of the Food and Drug Administration should conduct an annual risk assessment to determine whether the agency is conducting an adequate number of routine IRB inspections and to optimize the use of IRB inspections in the oversight of IRBs and protection of research participants. (Recommendation 3) |
In August 2023, HHS reported that FDA was taking steps to address this recommendation. Specifically, FDA planned to take steps to assess the number of annual IRB inspections, and, in May 2024, FDA completed this review. As part of this effort, FDA classified IRBs into organization types. FDA's assessment found that independent IRBs reviewed approximately half of FDA-regulated clinical research but represented less than 5 percent of IRBs selected for inspection in these fiscal years. FDA also assessed the feasibility of inspecting every IRB once every 5 years and determined it would require more than 300 routine inspections annually, which is 3 times the current number of IRB inspections conducted annually with existing resources. Given these findings, and the disproportionate amount of FDA-regulated research independent IRBs reviewed, FDA said they plan to inspect independent IRBs no less than once every 5 years and would not follow a set timeline for routine inspections of non-independent IRBs. In November 2025, FDA reiterated prior steps taken to address the recommendation and described other actions it is taking to optimize IRB inspections. In December 2025, the agency noted it was working to formally document the steps the agency would take annually in response to the recommendation. FDA clarified that it prioritizes high-volume IRBs, and uninspected IRBs and considers additional factors in determining which IRBs to inspect, such as time since last inspection and complaint history. FDA also noted it continues to work to systematically categorize IRBs into organization types, in part, to ensure the agency inspects high-volume independent IRBs at least once every 5 years. FDA reported it is fully implementing remote regulatory assessments, which began as a pilot program in fiscal year 2023, and is exploring the use of artificial intelligence tools with the goal of increasing the number of IRB inspections given their resources. Further, FDA and OHRP developed an information-sharing agreement to improve each agency's oversight capabilities. Based on the actions FDA has taken, we expect to close this recommendation as implemented upon review of FDA's revised procedures that document the actions FDA will take annually to determine whether the agency is conducting an adequate number of routine IRB inspections and to optimize the use of IRB inspections in the oversight of IRBs and protection of research participants.
|
| Department of Health and Human Services | The Secretary of Health and Human Services should ensure that OHRP and FDA convene stakeholders to examine approaches for measuring IRB effectiveness in protecting human subjects, and implement the approaches as appropriate. These could include effectiveness measures; peer audits of IRB meetings and decisions; mock protocols; surveys of IRB members, investigators, and human research participants; or other approaches. (Recommendation 4) |
HHS concurred with this recommendation, and has taken several actions that meet the intent of it. Specifically, OHRP requested that the Secretary's Advisory Committee on Human Research Protection (SACHRP) provide advice on IRB effectiveness, including the merits of potential effectiveness measures. In response, SACHRP convened meetings to discuss IRB effectiveness and approaches for measuring it in two public meetings held in 2023, and provided its recommendations to OHRP in October 2023. Additionally, HHS reported that the agencies obtained input from Common Rule departments and agencies and HHS Operating Divisions conducting or supporting human subjects research. OHRP and FDA also obtained input from various stakeholders at national and regional meetings. For example, in December 2023, the agencies gathered input on efforts to measure IRB effectiveness at a national conference of human subjects research oversight professionals. In October 2024, they convened a public meeting in which a panel of experts discussed four approaches for measuring IRB effectiveness. In convening stakeholders to discuss approaches for examining and ensuring IRB effectiveness, OHRP and FDA have acted on their responsibilities of protecting human research subjects. Although HHS did not implement new approaches at this time, they assembled experts who lead IRBs and human research protection programs to discuss a variety of options as well as the merits and limitations of them. For example, SACHRP made several recommendations, including using existing governmental and non-governmental standards and inspections and developing and sharing novel tools, and noted the importance of grant funding to assess the best methods and processes for measuring IRB effectiveness. Even in the absence of new approaches for examining IRB effectiveness, it will be important for HHS to continue to uphold its responsibilities in protecting human subjects.
|