Orphan Drugs: FDA Could Improve Designation Review Consistency; Rare Disease Drug Development Challenges Continue
Fast Facts
How do you get drug manufacturers to make drugs that treat rare diseases—when they may not sell enough to make a profit?
The Food and Drug Administration designates some drugs as "orphan drugs," which allows manufacturers to obtain incentives (like tax breaks) for developing drugs that treat rare diseases. But we found that FDA reviewers don’t always include all the required information in their reviews to determine whether a drug is eligible for orphan designation.
Especially as applications for orphan drug designations increase, we recommended improving how FDA reviews these applications.
Orphan Designation Applications, 2008-2017
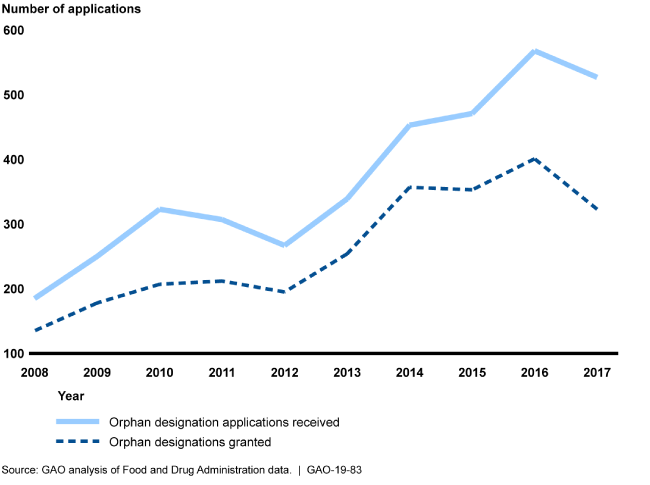
Line graph showing that orphan designation applications have nearly tripled since 2008.
Highlights
What GAO Found
The Food and Drug Administration's (FDA) Office of Orphan Products Development is responsible for reviewing drug manufacturer applications for orphan designation. Drugs granted this designation treat rare diseases and may receive various incentives under the Orphan Drug Act (ODA). As the number of orphan designation applications received and granted has grown, FDA outlined several process changes in its June 2017 modernization plan to improve designation review timeliness and consistency.
Orphan Designation Applications Received and Designations Granted from 2008 to 2017, as of April 2018
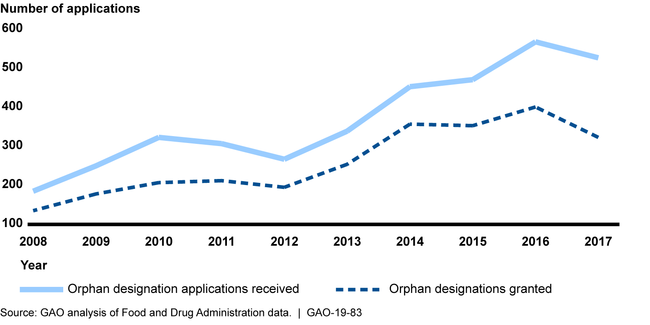
In evaluating designation applications, FDA reviewers generally apply two consistent criteria—(1) the size of the rare disease population, and (2) the scientific rationale that the drug may effectively treat the disease. To inform their evaluation, reviewers must record certain background information in a standard review template, such as the drug's U.S. marketing history. Officials told us this information provides important context, such as whether FDA has experience with a little known disease, critical to ensuring a complete designation application review. However, GAO's analysis of 148 designation review templates found that FDA does not consistently record or evaluate background information when making designation decisions. For example, 48 of 148 review templates GAO analyzed were missing information on the drug's U.S. marketing history. As such, FDA cannot be sure that reviewers are conducting complete evaluations that include all critical information needed for assessing its criteria.
Stakeholders GAO interviewed and research GAO reviewed identified a number of rare disease drug development challenges, such as the difficulty in recruiting small populations for clinical trials, with differing opinions about the ODA incentives. For example, several stakeholders were critical of manufacturers obtaining multiple orphan designations—and ODA incentives—for the same drug when the drug may otherwise be profitable from treating multiple patient groups. However, many patient advocacy groups noted that granting ODA incentives in these circumstances is needed to encourage drug manufacturers to study the safety and efficacy of drugs in rare disease populations.
Why GAO Did This Study
The ODA provides incentives, including tax credits and exclusive marketing rights, for manufacturers to develop drugs to treat rare diseases, which are typically defined as affecting fewer than 200,000 people in the United States. Approximately 7,000 rare diseases affect an estimated 30 million people in the United States, and only 5 percent of rare diseases have FDA-approved treatments.
GAO was asked to examine FDA's orphan drug processes. In this report, GAO examines, among other things, (1) the actions FDA has taken to address the growing demand for orphan designations; (2) the extent to which FDA has used consistent criteria and complete information in reviewing orphan designation applications; and (3) the steps FDA has taken to address rare disease drug development challenges. GAO analyzed FDA documents and data, as well as all designation review templates FDA completed as of March 2018 for applications received from October to December 2017. GAO interviewed agency officials, as well as stakeholders, including drug manufacturers, industry experts, and patient advocacy groups.
Recommendations
FDA should ensure that all required information for reviews of orphan designation applications is consistently recorded and evaluated. The agency concurred with our recommendation.
Recommendations for Executive Action
| Agency Affected | Recommendation | Status |
|---|---|---|
| Food and Drug Administration | The Commissioner of FDA should ensure that information from orphan drug designation applications is consistently recorded in Office of Orphan Products Development (OOPD) review templates and evaluated by OOPD reviewers when making an orphan designation decision. (Recommendation 1) |
In February 2019, FDA took action to address our recommendation by a) updating OOPD review templates and OOPD reviewer guidance for evaluating orphan designation applications, and b) training reviewers on these changes. For example, FDA added check boxes in the review template section on orphan drug designation history to confirm that there is no information to report for that section. In its guidance, FDA then further clarified the conditions under which a reviewer would select "none identified," such as when it is not clear if FDA has granted orphan designation for the same disease in the past. FDA also described the purpose of documenting orphan drug designation history in its guidance, which is to identify previous designations should any concerns arise with the application being reviewed.
|