Investigational New Drugs: FDA Has Taken Steps to Improve the Expanded Access Program but Should Further Clarify How Adverse Events Data Are Used
Fast Facts
FDA's expanded access program allows patients with serious or life threatening illnesses access to certain drugs before it has approved them. FDA also requires that manufacturers submit data about adverse reactions to these drugs.
While FDA has provided some guidance to manufacturers, FDA does not fully explain the few instances when it would use these data on adverse reactions. This may influence manufacturers' decisions to give these drugs to patients due to concerns that adverse reactions will result in FDA placing a hold on their drug.
We recommended that FDA clearly communicate how it uses these data in its drug approval process.
Types of expanded access that occur during the FDA's drug development/approval process
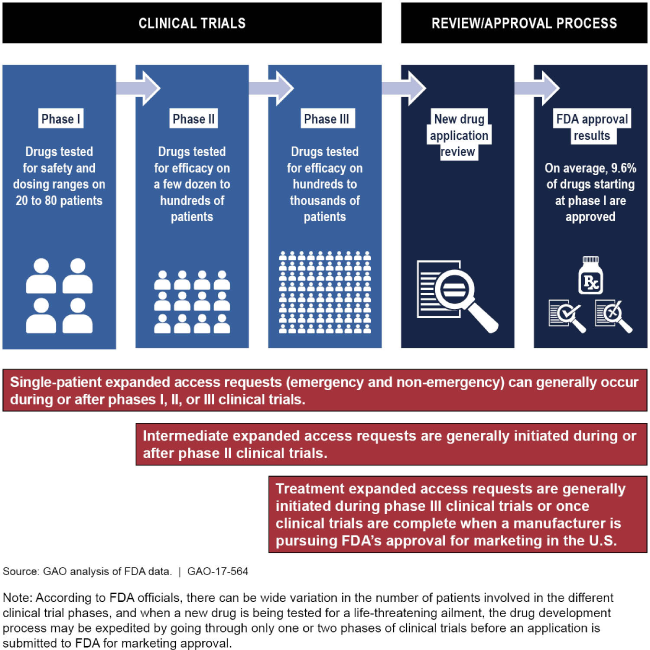
Graphic showing the stages of FDA's drug development/approval process.
Highlights
What GAO Found
Under the Food and Drug Administration's (FDA) expanded access program, patients with serious or life threatening ailments and no other comparable medical options can obtain access to investigational drugs outside of a clinical trial. Expanded access requests must be submitted to FDA but manufacturers must also grant permission for patients to access their investigational drugs. Of the nearly 5,800 expanded access requests that were submitted to FDA from fiscal year 2012 through 2015, FDA allowed 99 percent to proceed. Almost 96 percent of these requests were for single patients (either emergency or non-emergency). FDA's review process for expanded access requests is designed such that all requests are either allowed or not allowed to proceed within 30 days of receiving each request. FDA typically responded to emergency single-patient requests within hours and other types of requests within the allotted 30 days.
FDA and other stakeholders, including a non-profit organization and a drug manufacturer, have taken steps to improve the expanded access process and patient access to drugs. For example, in response to concerns that the process to request expanded access to drugs was complex and cumbersome, FDA simplified its website, guidance, and the forms required for the most common types of requests. Efforts by other stakeholders include a project to educate and streamline the process by which institutional review boards approve treatment plans for expanded access drug use and a pilot advisory group to help a drug manufacturer manage expanded access requests. Some states have also enacted “Right-to-Try” laws to facilitate patient access to investigational drugs. These laws provide liability and licensing protections for manufacturers and providers under state law if an adverse event—such as an adverse reaction to the drug—occurs with patients who were allowed access to investigational drugs. However, some stakeholders GAO interviewed cited concerns that these laws may not help patients access drugs, in part because they do not compel a manufacturer to provide access.
Manufacturers sponsoring clinical trials must submit safety reports to FDA that include adverse events data resulting from clinical trials and any expanded access use, to be used in assessing the safety of a drug within the drug approval process. FDA reported using these data from expanded access use in a few cases during the drug approval process but not more widely, because its use does not have the same controls as clinical trials. FDA officials reported that they communicate with manufacturers on how they will use expanded access adverse events data. However, GAO's review of documents FDA uses to communicate with drug manufacturers about the expanded access program found that only one included a reference to its use of these data, and it did not include specific examples of how the data might be used. Further, some of the manufacturers told GAO the guidance was unclear. These manufacturers noted that the lack of clear information can influence their decision whether to give patients access to their drugs because of their concerns that an adverse event will result in FDA placing a clinical hold on their drug, which could delay its development. This could impact FDA's goal of facilitating expanded access to drugs for treatment use by patients with serious or life-threatening diseases or conditions, when appropriate.
Why GAO Did This Study
FDA's goal for the expanded access program is to allow patients with immediately life-threatening and serious ailments access to investigational drugs when appropriate. Stakeholders have raised concerns that FDA's process is confusing or burdensome, particularly for the entities that have roles in the process—such as physicians, manufacturers, and institutional review boards.
GAO was asked to examine the expanded access program. Among other objectives, GAO examined 1) what is known about the number, type, and time frames of expanded access requests received by FDA; 2) what actions FDA and other stakeholders have taken to improve expanded access; and 3) how FDA uses data from expanded access in the drug approval process.
GAO reviewed regulations and FDA documents and analyzed FDA data on the numbers and types of expanded access requests it received from FY2012 through 2015, the most recent at the time of the review. GAO also interviewed FDA officials and other stakeholders including nine manufacturers—selected to represent large and small companies—and patient and physician representatives.
Recommendations
FDA should clearly communicate how it uses adverse events data from expanded access use in the drug approval process. FDA agreed with the recommendation.
Recommendations for Executive Action
| Agency Affected | Recommendation | Status |
|---|---|---|
| Food and Drug Administration | To help FDA meet its goal of facilitating expanded access to investigational drugs by patients with serious or life-threatening diseases or conditions, when appropriate, the Commissioner of FDA should clearly communicate how the agency will use adverse event data from expanded access use when reviewing drugs and biologics for approval for marketing and sale in the United States. |
In October 2017, FDA revised its guidance for industry entitled, "Expanded Access to Investigational Drugs for Treatment Use: Questions and Answers." In the revised guidance, FDA clarified how adverse event data from expanded access would be considered in the drug approval process and included a list of factors that reviewers of drugs and biologics consider when reviewing adverse event data from expanded access use. It also acknowledged that these factors make it difficult to link an expanded access treatment to a particular adverse event and noted that the agency is not aware of instances in which adverse events information from expanded access has prevented FDA from approving a drug.
|