Generic Drug User Fees: Application Review Times Declined, but FDA Should Develop a Plan for Administering Its Unobligated User Fees
Fast Facts
Nearly 90% of the prescription drugs dispensed in the United States are generics. The Food and Drug Administration must approve these drugs before they are marketed.
In 2012, a law allowed FDA to collect fees from drug manufacturers to support the review process. FDA committed to improving its process and meeting specific performance goals such as decreasing review times.
We found that FDA's reliance on user fees increased and that it surpassed many of its performance goals. However, we recommended that FDA make a plan for the fees it doesn't spend in the same year they're collected.
Generic Drug User Fee Collections, Obligations, and the Carryover for Fiscal Years 2013 through 2016
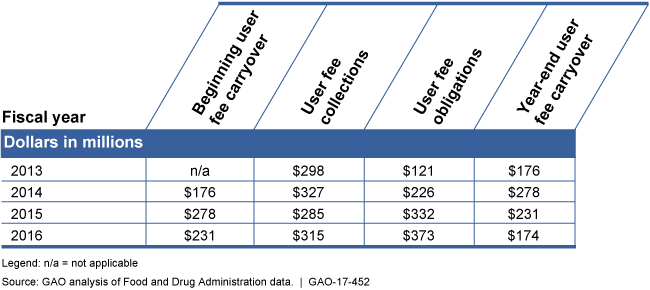
Table showing generic drug user fee collections, obligations, and carryover, 2013 - 2016.
Highlights
What GAO Found
Since the enactment of the Generic Drug User Fee Amendments of 2012 (GDUFA), the Food and Drug Administration's (FDA) reliance on user fees has increased from $121 million in fiscal year 2013 to $373 million in fiscal year 2016, or 45 percent of total program obligations in fiscal year 2013 to 76 percent in fiscal year 2016. FDA carried over $174 million in unobligated user fees at the end of the fourth year of the GDUFA 5-year period. GAO found that although FDA uses an internal management report to track user fee cash flows for internal purposes, it lacks a plan for administering its carryover—one that includes a fully-documented analysis of program costs and risks to ensure that program operations can be sustained in case of unexpected changes in collections or costs. GAO previously found that it is important for entities with carryover to establish appropriate target amounts based on program needs, risks, and contingencies. FDA's approach is inconsistent with best practices for managing federal user fees. Without a carryover plan, FDA lacks reasonable assurance that the size of its carryover is appropriate to ensure the efficient and responsible use of resources.
Generic Drug User Fee Collections, Obligations, and the Carryover, Fiscal Years 2013 through 2016
Dollars in millions
|
Fiscal year |
Beginning user fee carryover |
User fee collections |
User fee obligations |
Year-end user fee carryover |
|
2013 |
n/a |
$298 |
$121 |
$176 |
|
2014 |
$176 |
$327 |
$226 |
$278 |
|
2015 |
$278 |
$285 |
$332 |
$231 |
|
2016 |
$231 |
$315 |
$373 |
$174 |
Legend: n/a = not applicable
Source: GAO analysis of Food and Drug Administration data. | GAO-17-452.
FDA took steps to improve the timeliness and predictability of generic drug application reviews. FDA restructured the generic drug program by building a more robust organizational infrastructure, upgrading information technology systems, and implementing communication reforms. As FDA implemented these changes, it made additional refinements in response to applicants' feedback.
Generic drug application review times have improved under GDUFA. FDA's review time for a new generic drug application (known as an Abbreviated New Drug Application (ANDA)) decreased from 28 months for applications submitted in fiscal year to 2012 to about 14 months for those submitted in fiscal year 2015. FDA also surpassed multiple GDUFA performance goals. For example, FDA committed to reviewing 60 percent of ANDAs submitted in fiscal year 2015 within 15 months of their receipt. GAO found that FDA had taken action on 89 percent of such ANDAs for which it committed to conducting a substantive review by December 31, 2016, thereby surpassing this goal.
Why GAO Did This Study
Nearly 90 percent of prescription drugs dispensed in the United States are generic drugs. According to FDA, an increasing volume of generic drug applications over the past decades stressed its ability to review applications efficiently. GDUFA granted FDA the authority to collect user fees from the generic drug industry to supplement resources for the generic drug program. In return, FDA committed to meeting certain performance goals related to the timely review of generic drug applications and to implementing review process improvements.
GAO was asked to examine FDA's implementation of GDUFA. In this report, GAO (1) examines how user fees supported the generic drug program, (2) describes FDA's improvements to the generic drug application review process, and (3) analyzes changes in generic drug application review times. GAO reviewed laws and regulations; FDA policy, guidance, the GDUFA Commitment Letter, and GDUFA financial reports from fiscal years 2013 through 2016; FDA data on application review times from fiscal years 2012 through 2015; and interviewed officials from FDA, generic drug manufacturers, and trade associations.
Recommendations
GAO recommends that FDA develop a plan for administering user fee carryover that includes analyses of program costs and risks and reflects operational needs and contingencies. HHS agreed with GAO's recommendation.
Recommendations for Executive Action
| Agency Affected | Recommendation | Status |
|---|---|---|
| Food and Drug Administration | To ensure efficient use of generic drug user fees, facilitate oversight and transparency, and plan for risks, the Commissioner of FDA should develop a plan for administering user fee carryover that includes analyses of program costs and risks and reflects actual operational needs and contingencies. |
HHS concurred with this recommendation. In July 2019, the agency provided us with the fiscal year (FY) 2019 update to its five-year financial plan for the Generic Drug User Fee Amendments (GDUFA) program. The agency discussed the size of the FY 2018 user fee carryover balance and estimated collections, obligations, and carryover through FY 2022. The agency noted that it considers a reasonable carryover level to be large enough to provide for 8-10 weeks of resources to mitigate possible financial risks to the program, such as lower than expected user fee collections or a lapse in appropriations. The carryover at the end of FY 2018 exceeded this amount and the agency noted that it plans to reduce the carryover balance primarily through the hiring of term employees, which will provide additional resources to the program to help account for an unexpectedly high number of submissions. The agency plans to monitor carryover levels and adjust its plan as needed to ensure appropriate resource levels for the program. It also noted that it has made commitments to establish a resource capacity planning function and modernize its time report approach, which will provide the agency the ability to better forecast workload and translate forecasts into human resource and financial requirements. It has also made commitments to help enhance efficiency and transparency in the administration of GDUFA financial resources, including, as cited here, publishing a five-year financial plan and annual updates. Based on these actions, we are closing this recommendation as implemented.
|